TCBB 24 | 以集成方式充分利用受自然启发的计算方法进行癌症筛查 | Exhaustive Exploitation of Nature-inspired Computation for Cancer Screening in an Ensemble Manner
 记录一下我们在IEEE/ACM Transactions on Computational Biology and Bioinformatics上漫长的投稿旅程,21-Nov-2021 Submitted,26-Jul-2023 Major Revisions,07-Nov-2023 R1 Submitted,02-Apr-2024 Accept,时长总共约两年另四个半月,一如我的某段旅程曲折而漫长。论文信息如下:论文、代码
记录一下我们在IEEE/ACM Transactions on Computational Biology and Bioinformatics上漫长的投稿旅程,21-Nov-2021 Submitted,26-Jul-2023 Major Revisions,07-Nov-2023 R1 Submitted,02-Apr-2024 Accept,时长总共约两年另四个半月,一如我的某段旅程曲折而漫长。论文信息如下:论文、代码
一、摘要
癌症类型的准确筛查对于有效的癌症检测和精准的治疗选择至关重要。然而,基因表达谱与肿瘤之间的关联通常仅限于少数生物标记基因。虽然使用受自然启发的算法的计算方法在选择预测基因方面显示出潜力,但现有技术受到搜索效率低和跨多样化数据集的泛化能力差的限制。本研究提出了一种称为进化优化多样化集成学习(EODE)的框架,用于改善基因表达数据的癌症分类的集成学习。EODE方法结合了智能灰狼优化算法进行选择性特征空间缩减,引导随机注入建模进行集成多样性增强,以及子集模型优化进行协同分类器组合。在涵盖多种癌症类型的35个基因表达基准数据集上进行了大量实验。结果表明,与单个和传统聚合模型相比,EODE在筛查准确性上取得了显着改善。先进的特征选择、定向专业建模和合作分类器集成的综合优化有助于解决当前受自然启发的方法中的关键挑战。这为基于基因表达标志物的稳健和泛化的集成学习提供了有效的框架。
二、背景
癌症作为一种严重威胁人类健康的疾病,其发病率和死亡率在全球范围内持续攀升,给医疗卫生系统带来了沉重负担。准确及时地诊断癌症类型、评估病情进展和制定合理的治疗方案对于提高患者的生存率和生活质量至关重要。然而,由于癌症本身的异质性和复杂性,从分子水平上全面解析其发病机制并开发出有效的治疗手段仍然是一项极具挑战的任务。精准肿瘤学应运而生,它旨在利用患者的分子生物标志物(如基因、蛋白等)和多种组学数据,为每位患者量身定制个性化的治疗方案。其中,分析癌症患者的基因表达谱数据可以帮助我们识别与疾病发生发展密切相关的差异表达基因和调控通路,为开发分子诊断试剂、预后指标和药物靶点提供依据。尽管如此,要从高维度、稀疏和噪声较大的基因表达数据中准确检测出差异表达的生物标记物仍存在诸多分析挑战,如小样本量导致的统计力不足、技术噪声和批次效应的干扰、肿瘤细胞的遗传异质性以及患者之间的个体差异等,都会影响分析结果的可靠性。因此,发展出有效且稳健的计算分析方法来克服这些难题,从而精确鉴别不同癌症类型特异的分子生物标记物,对推进精准肿瘤学的发展具有重要意义。
三、动机
过去几年中,研究人员已经尝试运用机器学习、深度学习和多种生物启发式算法(如粒子群优化、蚁群优化、遗传算法等)来分析癌症基因表达数据并鉴定潜在的生物标记物,并取得了一定进展。然而,这些方法在实际应用中仍存在一些不足,如算法精度和稳健性有待提高、模型的泛化能力有限等。一个主要原因是它们大多依赖于单一的学习算法,而不同的算法在处理不同的癌症类型和数据集时,由于自身的偏置和局限性,很难保证能够始终获得最优性能。另一方面,单一学习器也难以充分挖掘数据中潜在的全部判别模式,降低了分析的鲁棒性。集成学习通过组合多个不同的基础学习器,可以有效克服单一模型的缺陷,发挥不同算法的长处,提高分类性能。然而,直接并列组合所有的基学习器并不是一种合理的策略,因为这会导致冗余表示的问题,即不同的基学习器可能会关注数据的相同特征,缺乏互补性,进而限制了整个集成系统的表达能力。近年来,研究人员开始探索如何利用智能优化算法来引导集成成员的选择,促进不同成员间的差异化和协同,但目前的方法还存在一些不足,如忽视了特征选择的重要性、无法充分挖掘数据的多样性等,制约了集成模型的性能发挥。因此,有必要设计出一种新颖的集成框架,将智能特征选择、引导多样性和元启发式优化等多种策略有机结合,来构建高性能的协同集成分类器,进一步推进癌症基因组学领域生物标记物的发现和精准肿瘤学的发展。

四、贡献
为解决上述挑战,本文提出了一种新的基于灰狼优化算法(GWO)的进化优化多样性集成学习框架(EODE)。EODE将GWO与包裹式特征选择、随机注入多样性增强和基学习器组合优化等策略紧密结合,以进一步提升集成分类器在癌症基因表达数据分析中的性能。具体来说,EODE首先利用GWO高效搜索高维基因表达空间,从中筛选出最具判别力的特征子集。这不仅降低了数据维度和噪声,也为后续机器学习分析创造了有利条件。其次,EODE通过随机化模型训练引入人为噪声,引导不同基学习器关注数据的不同方面,增强了集成成员的多样性。最后,EODE再次利用GWO优化基学习器的组合,在验证数据上评估每种集成方案,自动选择最优的集成。这种策略可最大化不同基学习器的协同作用,提高集成系统的泛化能力。我们在35个癌症基因表达数据集上评估了EODE,结果显示其在分类精度、平均性能和特征子集大小等指标上均显著优于23种现有方法,充分验证了该框架的有效性和优越性。EODE通过GWO驱动的特征选择、多样性引导和集成优化,不仅推进了生物标记物发现,也为精准肿瘤学的发展注入了新的活力。
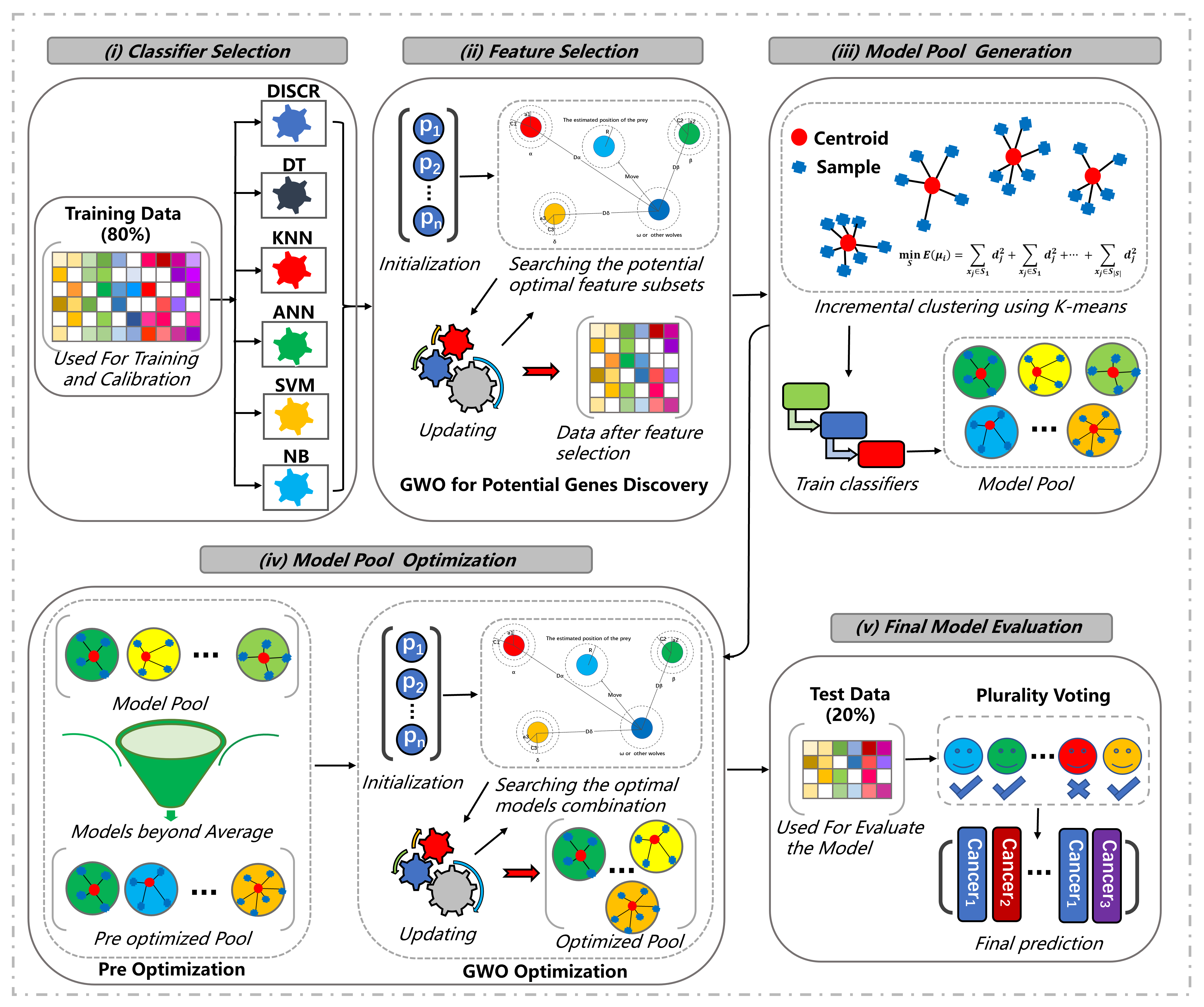
EODE算法概述(图1):在灰狼优化器(GWO)特征选择阶段,原始的癌症基因表达训练数据被用来训练所有基分类器,并选择性能最佳的分类器作为评估分类器。然后对处理过的数据进行优化,构建集成模型。具体而言,训练数据使用K均值方法逐步进行聚类,形成子空间聚类。这些聚类用于训练各个基分类器,然后将其添加到模型池中。模型池中任何表现低于平均水平的分类器都将被过滤掉。接下来,应用GWO对分类器池进行优化,并确定最佳的集成组合。最后,使用多数投票策略在独立测试数据集上评估优化后的集成模型,生成最终的癌症类型预测结果。
五、EODE方法
5.1. 方法概述
本研究提出了一种名为EODE的新型自然启发式方法,用于快速识别多种癌症类型的生物标记基因。算法的整体框架如图1所示。输入为原始基因表达数据$D_{or} = |{(x_1,y_1),…,(x_n,y_n)|}$,其中$x_i$表示一个包含$dim$个基因的样本,$y$为分子亚型标签,$n$为总样本数。在特征选择步骤中,我们利用GWO在训练集$D_{tr}$上训练模型,从中提取出相关的生物标记基因。首先,从基学习器池$B$(包括判别分析、决策树、K近邻、人工神经网络、支持向量机和朴素贝叶斯)中选择表现最佳的分类器作为特征选择的评估器。随后,我们利用筛选出的特征训练和优化一个多样性集成模型$\Psi$。具体来说,数据首先通过5折交叉验证进行处理。然后,我们使用K-means方法将数据划分为多个子空间,并在这些子空间上训练基学习器,组成模型池。对于池中表现较差的模型,我们将其过滤掉。最后,我们应用GWO优化模型池,找到最佳的模型组合。在测试集上,我们采用多数投票的方式评估最终模型$\Psi$的性能。EODE的整体框架如算法1所示。该方法充分利用GWO的特征选择能力和集成优化策略,在多个癌症基因表达数据集上展现出显著的性能优势,为生物标记基因的快速发现和精准肿瘤学的应用提供了新的解决方案。

5.2. 受自然启发的特征选择
考虑一个训练癌症基因表达数据 \(D_{tr} = \{(x_1,y_1),...,(x_n,y_n)\}\),其中 \(x_i = (x_{i,1},x_{i,2},...,x_{i,dim})\) 表示特征向量,$dim$ 表示特征的数量,$y$ 属于集合 \(\{1,2,..,c\}\) 表示类别,$n$ 是样本数量。需要注意的是,基因表达数据的高维特性可能包含许多不相关的基因,这可能会对识别准确性产生负面影响,同时增加计算时间。因此,进行特征选择对有效预处理数据至关重要。
灰狼优化器 (GWO),最初由Mirjalili提出,是一种受自然界灰狼社会等级和狩猎行为启发的群体智能算法。GWO具有收敛速度快、参数调整少、易于实现等优点。GWO的核心概念围绕着三种主要的捕食行为:围捕猎物、狩猎和攻击猎物,这些行为基于灰狼之间的社会等级进行。GWO中的社会等级分为四个级别:$\alpha$、$\beta$、$\delta$ 和 $\omega$,其中 $\alpha$ 是主导的狼,接着是 $\beta$ 和 $\delta$,而剩下的狼被标记为 $\omega$。等级较高的狼对等级较低的狼施加支配,$\alpha$、$\beta$ 和 $\delta$ 在算法中发挥关键作用,$\alpha$ 是狼群之王,$\beta$ 和 $\delta$ 则作为潜在的接班人。$\alpha$ 狼代表最适合的解决方案,并引导着狼群朝着有前景的搜索区域前进。第二和第三最适合的解决方案被建模为 $\beta$ 和 $\delta$ 狼。$\omega$ 狼代表剩余的较弱候选解决方案,它们遵循 $\alpha$、$\beta$ 和 $\delta$ 狼的指导。在优化过程中,候选解决方案会迭代地向最佳的三个解决方案更新其位置,直至收敛于全局最优值。具体来说,GWO的示意图如图2所示。
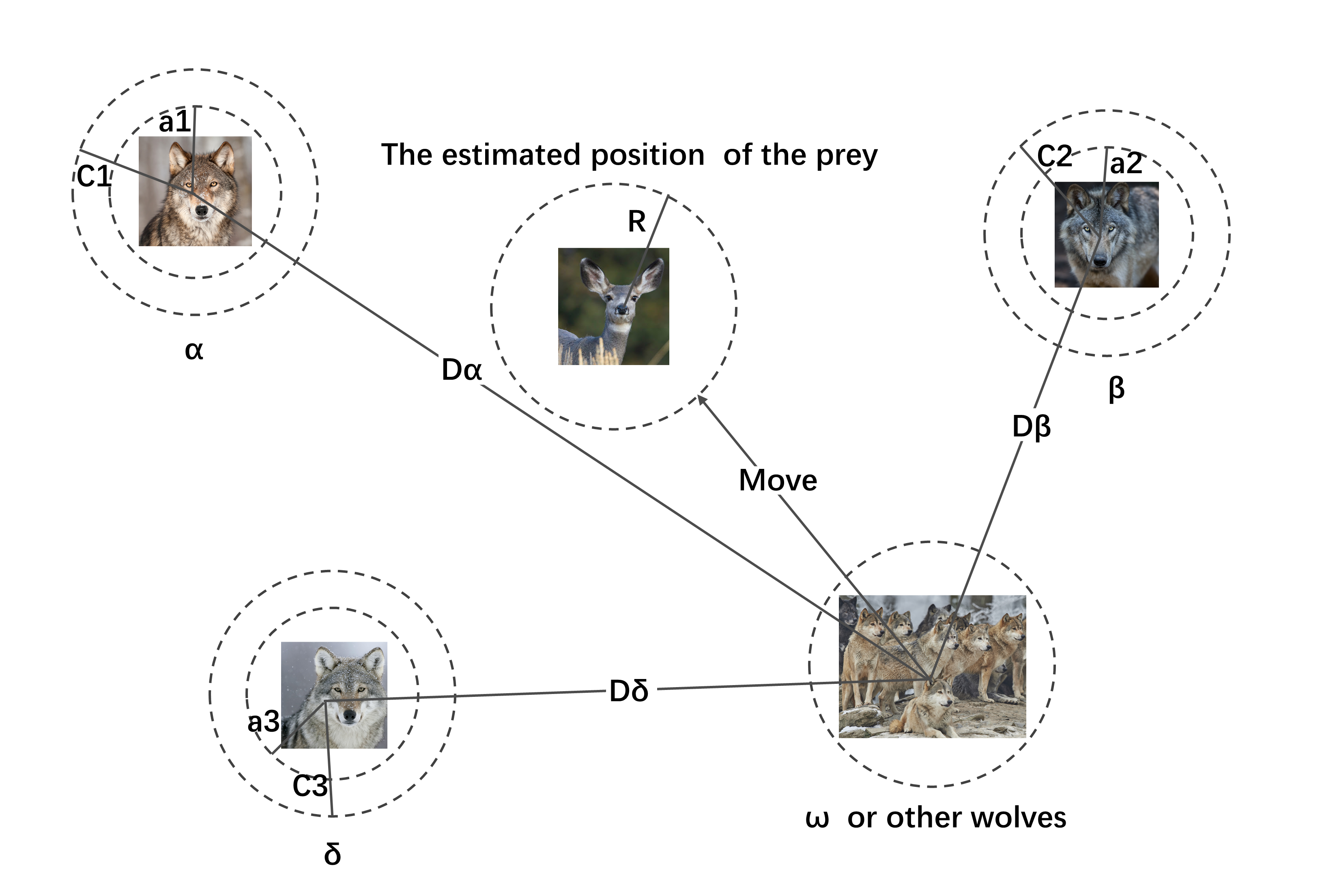
狼群位置的更新过程(图2):首先,狼群的位置在解空间内随机初始化。然后,根据适应度函数评估每只狼的适应度。在每一次迭代中,狼群的位置会根据数学公式进行更新,考虑了狼群的社会等级,其中 $\alpha$ 狼具有最大的影响力。更新过程中,其他狼会被吸引朝向 $\alpha$、$\beta$ 和 $\delta$ 狼的位置移动。这种迭代式的位置更新会持续进行,直到满足终止条件。最终,$\alpha$ 狼的位置代表了GWO算法找到的最佳解决方案。
基于这些基础,我们提出了一种基于GWO的受自然启发的特征选择方法,包括六个基本组成部分:分类器选择、种群初始化、围捕猎物阶段、狩猎阶段、攻击阶段和特征选择目标函数。
5.2.1. 分类器选择
为了评估特征选择结果,我们在分类器池 $B$ 中考虑了六个基本分类器:判别分析(DISCR)、决策树(DT)、K-最近邻(KNN)、人工神经网络(ANNs)、支持向量机(SVM)和朴素贝叶斯(NB)。然而,在特征选择阶段将所有这些分类器都纳入集成方法中会带来计算上的昂贵。因此,我们采用预训练方法来从池 $B$ 中选择性能最佳的分类器。癌症基因表达数据 $D$ 在每个基分类器上进行了五折交叉验证,性能最好的分类器被选为特征选择阶段的评估分类器。这种方法使我们能够有效地选择最适合的分类器,用于后续的特征选择过程。
5.2.2. 种群初始化
初始时,种群 $\vec{X}$ 被随机创建并表示为实数。每个个体,表示为 $\vec{X_i}$,是一组基因:\(\vec{X_i} = \{g_1, g_2, ..., g_{dim}\}\),其中 $g_{dim}$ 表示第 $dim$ 个基因,$dim$ 是基因总数。为了将这些实数转换为二进制形式,我们使用一个阈值 $\theta$。如果一个特征值 ($g_n$) 大于或等于 $\theta$,则设为 1,表示选择了相应的特征。另一方面,如果 $g_n$ 小于 $\theta$,则设为 0,表示未选择该特征。之后,每个个体的位置由一个二进制(0/1)字符串表示。
5.2.3. 围捕猎物阶段(Encircling Prey Phase)
“围捕猎物” 行为是灰狼群用来搜索特征子集的策略。该行为在数学上被建模,以模拟灰狼逐渐接近并包围猎物的过程。灰狼与猎物之间的距离 ($\vec{D}$) 由以下方程确定:
\[\begin{equation} \vec{D} = |2 \cdot r_2 \cdot \vec{X_p(t)} - \vec{X_i(t)}|, \end{equation}\]其中 $\vec{D}$ 表示它们之间的距离。在搜索过程中,当前迭代由 $t$ 表示,$\vec{X_p(t)}$ 和 $\vec{X_i(t)}$ 分别表示猎物和灰狼的位置向量。
为了更新灰狼的位置,我们利用公式 $ \vec{X_i(t+1)} = \vec{X_p(t)} - (2\vec{a} \cdot r_1 - \vec{a}) \cdot \vec{D}$。这里,$\vec{a}$ 是收敛因子,随着迭代的进行线性减小从 2 减小到 0。收敛因子的计算方式为 $\vec{a} = 2 - 2t$/$max_t$,其中 $t$ 表示当前迭代次数,$max_t$ 是定义搜索过程的最大迭代次数。此外,$r_1$ 和 $r_2$ 是介于 0 和 1 之间的随机数。通过应用这个位置更新公式,灰狼调整其位置朝着猎物。这里$(2\vec{a} \cdot r_1 - \vec{a})$ 决定了移动的大小和方向,而距离 $\vec{D}$ 指导了灰狼在缩小与猎物的距离方面的移动。该过程迭代进行,直到达到所需的最大迭代次数 $max_t$。最终,灰狼通过围捕猎物,获得一个富含信息的特征子集。
5.2.4. 狩猎阶段(Hunting Phase)
灰狼具有识别猎物大致位置并协作将其包围的能力。然而,在许多未知情况下,它们可能不会精确了解目标的确切位置。在我们的研究中,通过引入三个关键个体:$\alpha$、$\beta$ 和 $\delta$,模拟了灰狼的行为。这些个体有助于引导整个狼群围捕猎物并搜索最佳解决方案。为了追踪猎物的位置,通过下述公式计算每只灰狼到猎物的距离:
\(\begin{equation} \vec{D}_\alpha = |\vec{C}1 \cdot \vec{X}\alpha - \vec{X}i|, \end{equation}\) \(\begin{equation} \vec{D}\beta = |\vec{C}2 \cdot \vec{X}\beta - \vec{X}i|, \end{equation}\) \(\begin{equation} \vec{D}\delta = |\vec{C}3 \cdot \vec{X}\delta - \vec{X}_i|. \end{equation}\)
其中,$\vec{D_{\alpha}}$、$\vec{D_{\beta}}$ 和 $\vec{D_{\delta}}$ 分别表示灰狼 $\alpha$、$\beta$、$\delta$ 与猎物之间的距离。$\vec{X_{\alpha}}$、$\vec{X_{\beta}}$ 和 $\vec{X_{\delta}}$ 表示 $\alpha$、$\beta$ 和 $\delta$ 的位置,而 $\vec{X_i}$ 表示灰狼当前的位置。此外,$\vec{C_1}$、$\vec{C_2}$ 和 $\vec{C_3}$ 是用于计算这些距离的随机向量。
每只灰狼根据这些距离计算更新其位置: \(\begin{equation} \vec{X}' = \vec{X}\alpha - A_1 \cdot \vec{D}\alpha, \end{equation}\) \(\begin{equation} \vec{Y}' = \vec{X}\beta - A_2 \cdot \vec{D}\beta, \end{equation}\) \(\begin{equation} \vec{Z}' = \vec{X}\delta - A_3 \cdot \vec{D}\delta. \end{equation}\)
这里,$\vec{X}’$、$\vec{Y}’$ 和 $\vec{Z}’$ 分别表示向 $\alpha$、$\beta$ 和 $\delta$ 移动的灰狼的新位置。常数 $A_1$、$A_2$ 和 $A_3$ 控制向猎物移动的幅度。
最后,灰狼在下一个时间步的位置 $\vec{X_i(t+1)}$ 用位置 $\vec{X}’$、$\vec{Y}’$ 和 $\vec{Z}’$ 的平均值来表示: \(\begin{equation} \vec{X_i(t+1)} = \frac{\vec{X}' + \vec{Y}' + \vec{Z}'}{3}. \end{equation}\) 通过这种方式,整个狼群一起朝着 $\alpha$、$\beta$ 和 $\delta$ 的位置移动,每个个体的新位置随之更新。
5.2.5. 攻击阶段(Attacking Phase)
捕猎过程的最后阶段是攻击阶段,在此期间,灰狼旨在捕捉猎物并获得最佳解决方案。该阶段涉及调整某些参数以在全局探索和局部利用之间取得平衡。为了实现这种平衡,考虑了两个关键参数:$a$ 和 $A$。参数 $a$ 的值以线性方式从 2 逐渐减小到 0。同时,$A$ 的波动范围也在减小。参数 $A$ 的值在区间 $[-a, a]$ 内取值。灰狼的行为受 $A$ 的大小影响。当 $A$ 的绝对值大于 1 时,灰狼倾向于分散到不同区域,实现对猎物的全局搜索。相反,当 $A$ 的绝对值小于 1 时,灰狼表现出更专注的局部搜索。
除了这些参数之外,灰狼位置对猎物的影响受到随机权重的控制,记为 $C$。该权重在 0 到 2 之间变化,决定了灰狼位置对猎物的随机影响。大于 1 的 $C$ 值表示更高的权重,强调了灰狼位置在指导搜索中的重要性。相反,小于 1 的 $C$ 值分配了较低的权重,减少了灰狼位置的影响。这种随机权重 $C$ 有助于防止算法过早收敛并陷入局部最优解。通过在攻击阶段动态调整 $a$、$A$ 和 $C$ 的值,灰狼在探索和利用之间取得平衡,使其能够有效地搜索和捕获最佳解决方案,同时避免过早收敛和局部最优解。
5.2.6. 特征选择的目标函数
在 GWO 算法的每次迭代中,对于每个候选解 $\vec{X_i}$,都使用从分类器选择阶段选定的评估分类器来预测其分类标签。具体而言,评估分类器最初在原始的训练基因表达数据集 $D_{tr}$ 上,利用五折交叉验证使用所有特征进行训练。对于每个包含一组所选特征的 $\vec{X_i}$,评估分类器通过仅使用 $\vec{X_i}$ 中的所选特征对 $D_{tr}$ 中的相应数据点进行分类,生成预测标签 $y_i’$。$y_i’$ 在 $D_{tr}$ 上的表现确定了分配给解 $\vec{X_i}$ 的适应度值。这使得 GWO 算法能够确定 $\alpha$、$\beta$ 和 $\delta$ 解,这些解代表了当前最佳的特征子集以用于分类。
在特征选择阶段,主要目标是在癌症基因表达数据中识别和选择相关特征,并滤除冗余特征,以供后续识别使用。传统研究通常仅关注分类准确性,忽略了与冗余特征相关的资源成本。在我们的研究中,我们通过将分类准确性和特征子集大小视为特征选择目标函数的一部分来解决这一局限性。特别地,目标函数被记为 $f_1$,定义如下: \(\begin{equation} f_1 = \alpha * \text{error} + \beta * \frac{f_{\text{num}}}{\text{dim}}. \end{equation}\)
这里,$f_{\text{num}}$ 表示进化过程中所选择特征的数量,$\text{dim}$ 表示数据集中特征的总数。为了在两个目标之间取得平衡,我们引入权重系数来控制它们的相对重要性。在我们的研究中,$\alpha$和 $\beta$ 的权重分别被设置为 0.9 和 0.1。这些权重系数是基于参考文献中的研究结果确定的,其中分类准确性被确定为主要目标。
分类误差($\text{error}$)是目标函数的一个关键组成部分。它被计算为 1 与准确率($\text{acc}$)的差值,准确率定义如下: \(\begin{equation} \text{error} = 1 - \text{acc}, \end{equation}\) \(\begin{equation} \text{acc} = \frac{\sum_{s=1}^n{I(y'_s,y_s)}}{n}. \end{equation}\)
5.3. 受自然启发的多样化集成学习
在本节中,我们提出了一种新颖的自然启发的多样化集成学习方法,通过自然启发的特征选择得到的选定特征,来提高癌症识别的性能。我们的方法包括多样化子空间生成、模型池生成和模型池优化。
5.3.1. 多样化子空间生成
给定特征选择后的基因表达数据,表示为 \(D_{tr}'=\{(x_1,y_1),...,(x_m,y_m)\}\),其中 $x_i =(x_{i,1}, x_{i,2}, …, x_{i,dim})$ 表示具有 $dim$ 个特征的特征向量,\(y \in \{1,2,...,c\}\) 表示分类标签,$m$ 表示输入样本的数量,我们使用 K-means 方法将输入的癌症基因表达数据聚类成多个簇。聚类过程从 1 到 $t$ 迭代进行,每次迭代生成 $K$ 个簇。这里,$t$ 表示迭代的总次数。通过最小化以下函数获得簇: \(\begin{equation} \mathop{argmin}\limits_S \sum_{i=1}^K\sum_{x \in S_i}{||x - \mu_i||^2}, \end{equation}\) 其中 $x$ 表示特征向量,$\mu_i$ 是簇 $S_i$ 的中心点。这个聚类过程生成了一组由所有获得的簇组成的多样化子空间。
5.3.2. 模型池生成
每个多样化子空间中的簇用于训练六个分类器(DISCR、DT、KNN、ANN、SVM 和 NB),以创建一个模型池。在此步骤中使用的基础分类器是独立的。得到的模型然后被添加到基础模型池 $\mathcal{MP}$ 中。基础模型池 $\mathcal{MP}$ 包括 $l * |\mathcal{B}|$ 个模型,其中 $l$ 表示簇的数量,$|\mathcal{B}|$ 表示基础分类器的数量。最后,我们使用自然启发的优化技术来改进集成中的基础模型。在这里,可以利用任何分类器的组合。
5.3.3. 模型池优化
在获得多样化的基础模型池 $\mathcal{MP}$ 后,我们提出了一个预优化步骤,通过移除表现低于平均水平的模型来优化 $\mathcal{MP}$。随后,我们采用自然启发的优化方法,即 GWO,来进一步优化经预优化的基础模型池 $\mathcal{MP}$。
5.3.3.1 种群初始化
种群是随机初始化的,每个个体表示如下:
\[\begin{equation} \vec{X_i} = \{mp_1, mp_2,..., mp_r\}. \end{equation}\]这里,$mp_r$ 表示模型池 $\mathcal{MP}$ 中的一个分类器,$r$ 是 $\mathcal{MP}$ 中模型的总数。与自然启发的特征选择类似,模型的选择或非选择由二进制值表示。“1” 表示选择了模型,而 “0” 表示没有选择模型。为了将 GWO 的连续搜索空间转换为二进制搜索空间,我们引入了一个阈值 $\theta$,大于 $\theta$ 的值被转换为1,反之转换为0。
5.3.3.2 自然启发的优化过程
在这个阶段,我们的目标是通过优化基础模型池 $\mathcal{MP}$ 来发现最佳的模型子集。种群用于在包围阶段中探索最佳模型子集,在捕猎阶段中识别潜在的最佳解决方案,并最终在攻击阶段获得最佳解决方案。
5.3.3.3 集成优化目标函数
我们的目标是在最小的集成大小下实现最高的识别性能。在特征选择后对数据进行聚类和训练基础分类器以创建模型池后,我们的目标是优化模型池,以获得具有最小大小的最佳集成模型。优化后的模型集合然后使用测试数据进行评估。模型池优化阶段的目标函数,记为 $f_2$,定义如下: \(\begin{equation} f_2 = \alpha * \text{error} + \beta * \frac{|\psi|}{r}. \end{equation}\)
这里,$\text{error}$ 表示方程 (10) 中描述的识别错误率,\(\psi\) 的绝对值是选定模型的总数,$r$ 是 $\mathcal{MP}$ 中的模型数量。$\alpha$ 和 $\beta$ 的设置与第 5.2.6 节中相同,其中 $\alpha$ 占 90% 的重要性,$\beta$ 占 10%。然而,与第 5.2.6 节不同的是,这里的预测标签 $y’_s$ 不是由单个分类器预测的,而是考虑了多个模型的集成。我们采用多数投票方法来组合多个模型的预测,这在许多研究中已被证明是一种简单有效的集成融合技术。
5.3.3.4 集成分类器预测
在训练过程中,我们获得多个模型 $\psi$ 来表示模型 $\Psi$。模型 $\Psi$ 被用于生成一个集成,所有模型在 $\Psi$ 中都被用于预测测试集。利用多数投票方法融合模型 $\Psi$ 中所有模型的预测类标 $y’_s$。识别准确率可以使用公式 (11) 计算。
六、实施细节
6.1. 数据集
本文研究所用的癌症基因表达数据集是可从网站下载。这 35 个数据集涵盖了多种癌症类型,具有超过 1000 个维度的高维特征,但样本量相对较小(如TABLE 1所示)。这给了我们一个“维度灾难”的挑战,需要开发出具有高鲁棒性和良好泛化能力的计算模型,以应对不同类型的癌症。
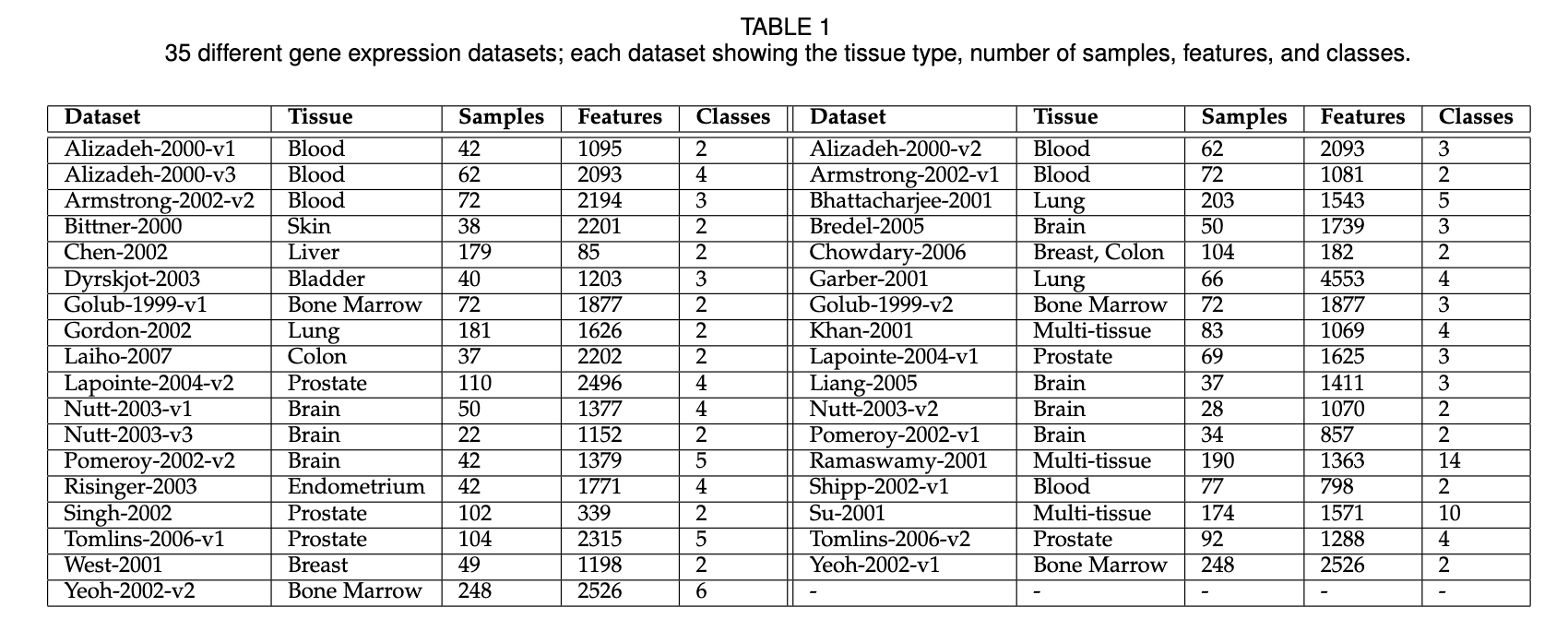
为了进行严格的评估,我们在实验开始前将这些原始数据集随机分成了不相交的训练集和测试集,比例为 80:20。其中,训练集占据了数据的 80%,用于模型训练和超参数调优;而测试集则占据了数据的 20%,仅用于最终评估完全训练模型的性能。这样做可以确保对泛化能力的无偏估计。我们在进行精确的训练/测试拆分时保持了每个集合中类的平衡。具体的训练和测试数据集可以从我们的代码库下载。在模型选择和超参数调优阶段,我们仅在训练数据上使用了 k 折交叉验证(k=5)。通过这种严格的方法,我们可以评估跨多种癌症类型的真实泛化误差和鲁棒性。
6.2. 基准方法
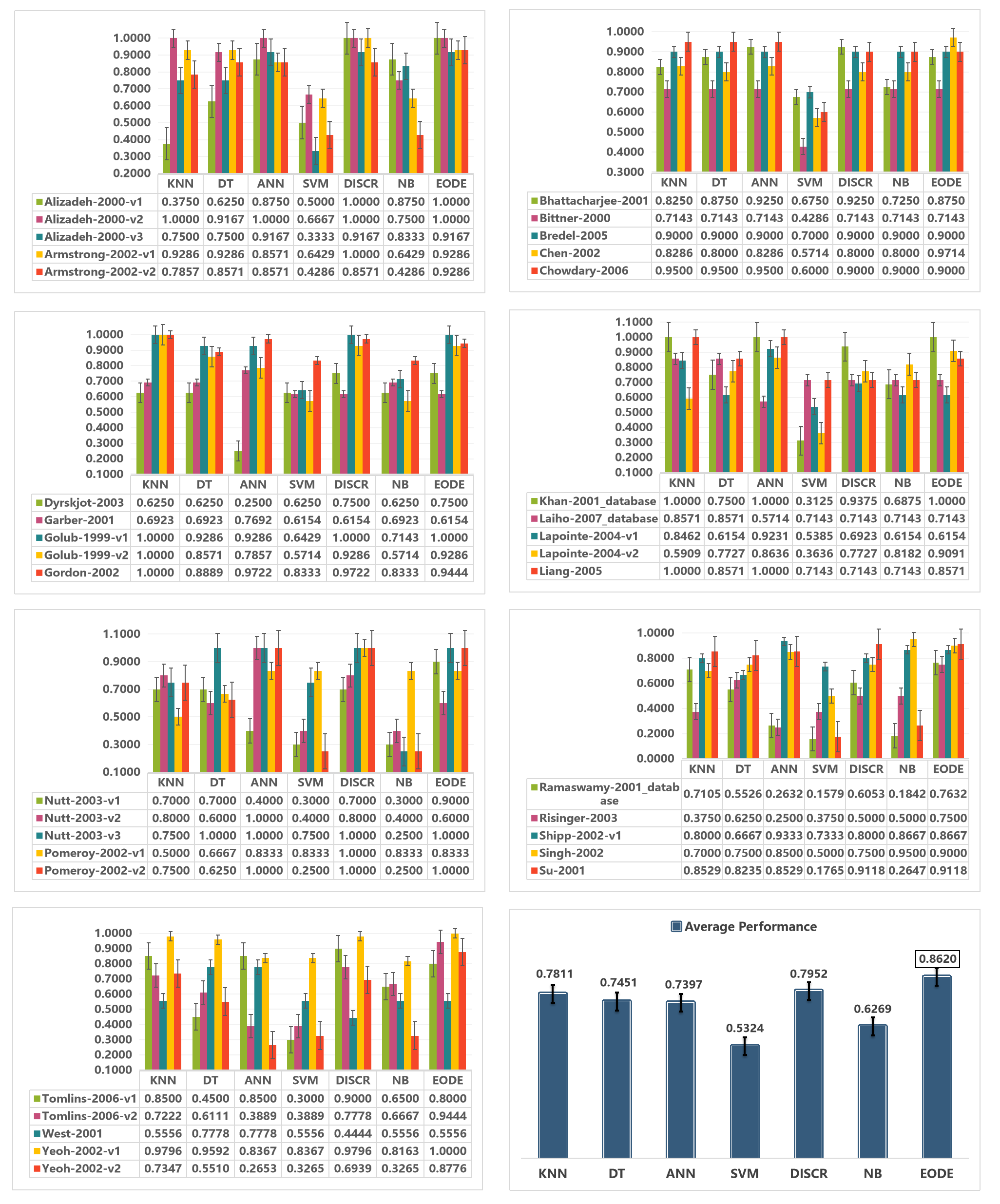
为了评估我们提出的方法的有效性,我们将其与文献中广泛使用的几种现有分类器和集成算法进行了比较。首先,我们将我们的模型与六个基本分类器进行了比较:DISCR(判别分析)、DT(决策树)、KNN(K近邻)、ANN(人工神经网络)、SVM(支持向量机)和NB(朴素贝叶斯)。这些分类器作为性能比较的基线。
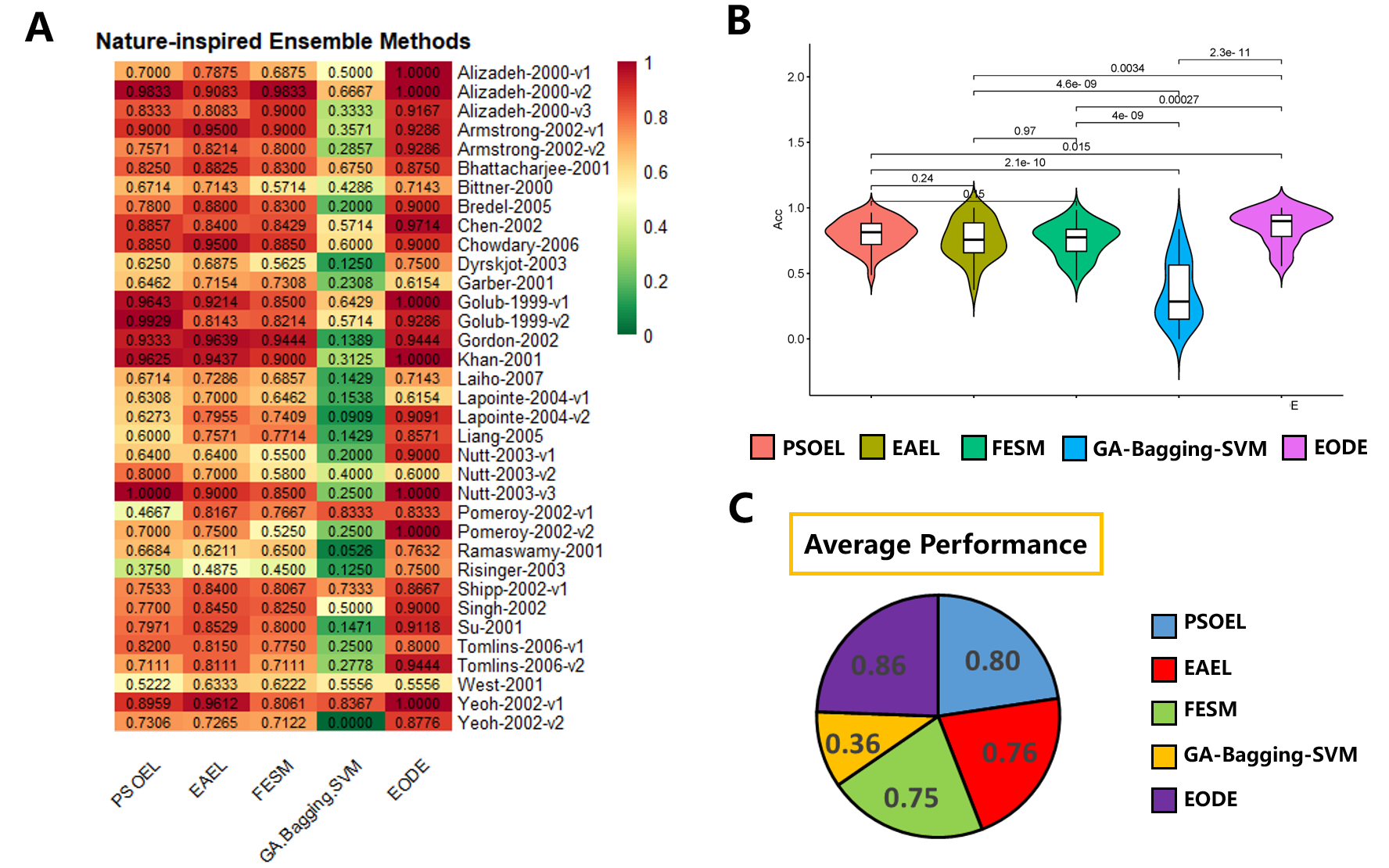
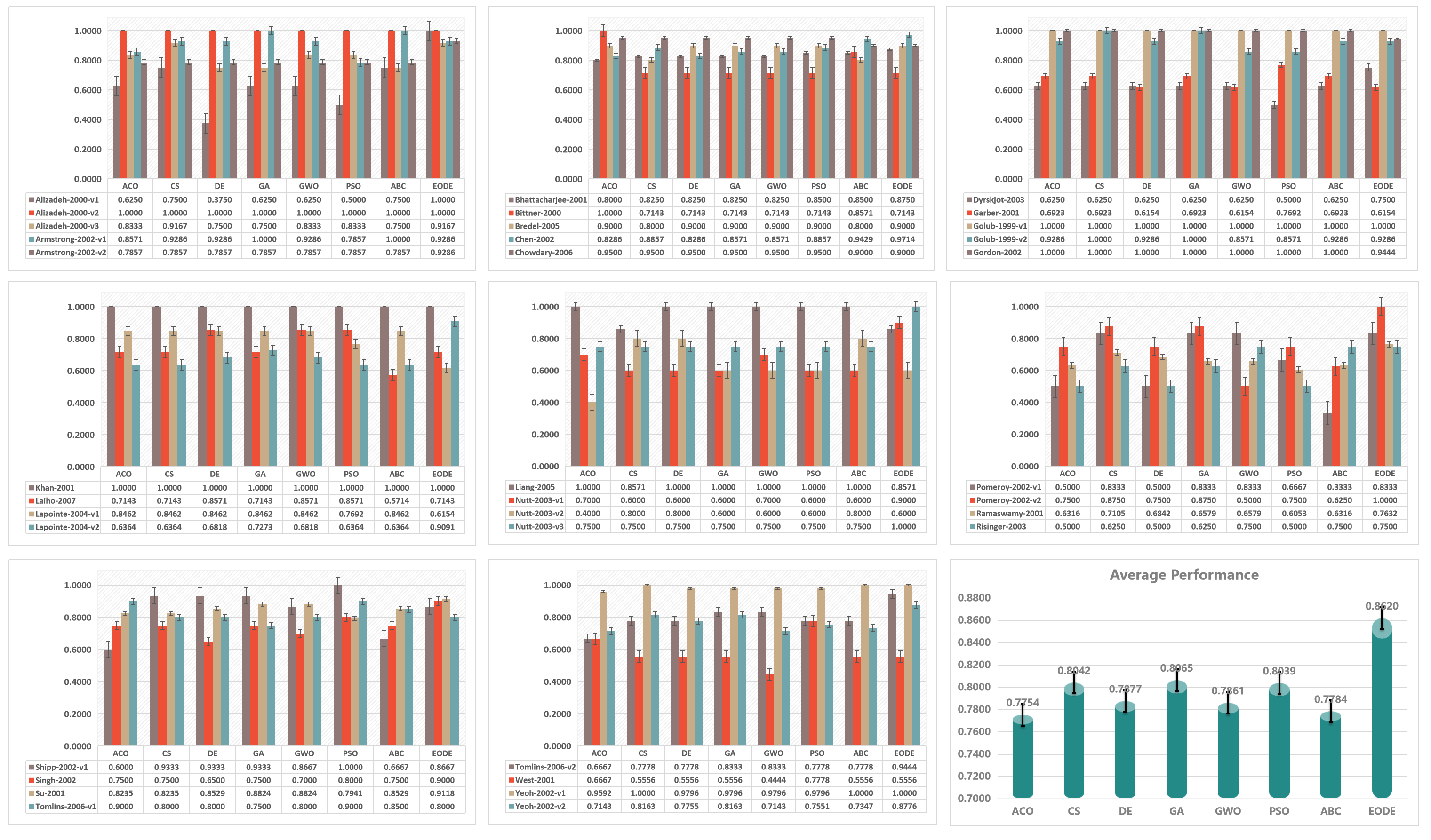
接下来,我们将我们的方法与七种进化算法进行了比较:ACO(蚁群算法)、CS(布谷鸟搜索算法)、DE(差分进化算法)、GA(遗传算法)、GWO(灰狼优化算法)、PSO(粒子群算法)和ABC(人工蜂群算法)。这些算法被广泛应用于优化问题。此外,我们还将我们的方法与四种新型集成方法进行了比较:PSOEL 、EAEL 、FESM 和GA-Bagging-SVM 。选择这些方法是为了展示我们提出的方法相对于集成学习中的最新进展的有效性。
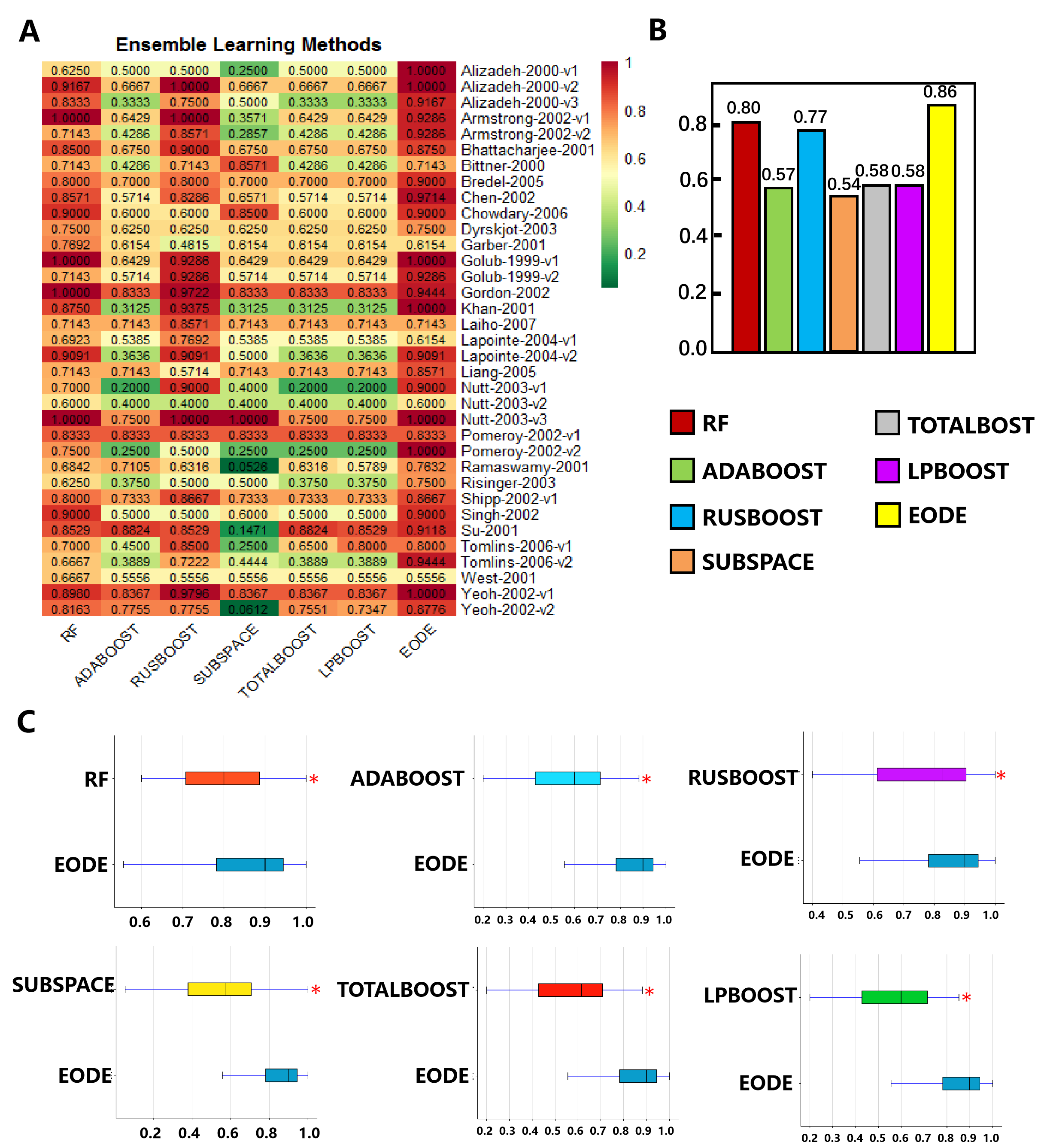
此外,我们将我们的集成算法与六种集成分类器进行了比较:随机森林(RF)、ADABOOST(自适应增强)、RUSBOOST(随机欠采样增强)、SUBSPACE(子空间集成)、TOTALBOOST(全局增强)和LPBOOST(最小边界增强)。通过将我们的方法与这些多样化的算法进行比较,我们旨在展示其在解决癌症基因表达数据分类问题方面的有效性。
6.3. 实验设置
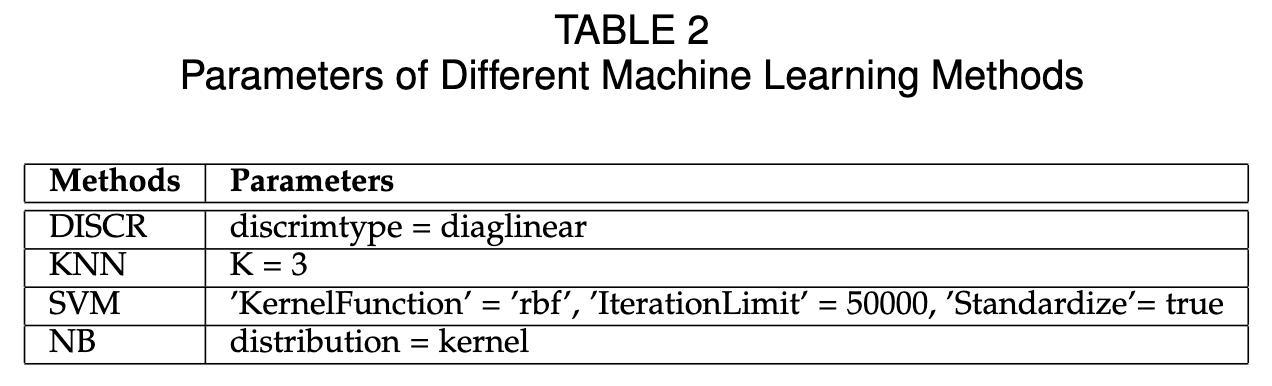
我们用以实验的台式机的参数和使用的软件版本信息如下所示:Intel(R) Core(TM) i7-10700KF CPU @3.80GHz,32GB的内存,以及64位的Windows 10操作系统,使用Matlab 2021a。我们利用了六个基本分类器,分别是DISCR、DT、KNN、ANN、SVM和NB,构建了集成模型。DISCR、KNN、SVM和NB的参数总结在TABLE 2中,而其余分类器则采用默认设置。此外,随机森林(RF)、ADABOOST、RUSBOOST、SUBSPACE、TOTALBOOST和LPBOOST均采用默认参数值。此外,四种新型集成分类器方法,即PSOEL、EAEL、FESM和GA-Bagging-SVM的参数设置与原始论文保持一致。
在我们的实验中,原始数据以8:2的比例被随机分为训练集和测试集。同时,五折交叉验证方法被使用对数据进行训练。对于特征选择和集成优化中的GWO算法,其种群大小($P$)为100,迭代次数为50,阈值($\theta$)为0.5。特别地,在我们的研究中,阈值 $\theta$ 用以灰狼优化算法中作为特征选择的判断标准,并不与实际基因表达值相关。在聚类阶段,参数$t$被设置为$\sqrt[5]m$。七种经典进化算法的详细参数,包括ACO、CS、DE、GA、GWO、PSO和ABC,总结在TABLE 3中,其中种群大小($P$)和最大迭代次数($max_t$)设置为相同的值。 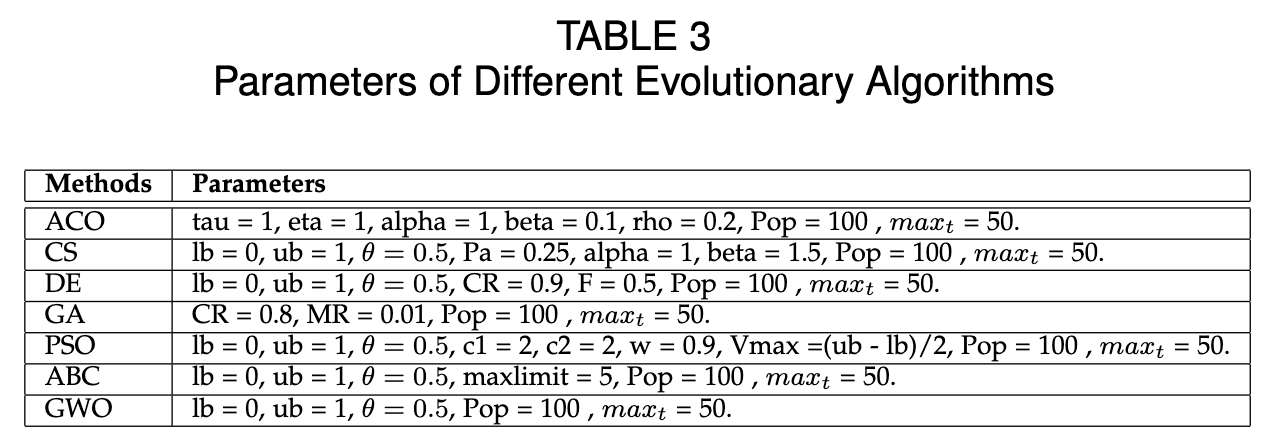
七、结果与分析
7.1. 与其他受自然启发的集成学习算法的性能比较
7.2. 与不同机器学习算法的性能比较
7.3. 与不同进化计算方法的性能比较
7.4. 与不同集成学习方法的性能比较
八、结论与未来工作
癌症类型的识别对于癌症研究至关重要,因为它可以实现早期诊断和个性化治疗。在这个领域,一个关键的挑战是识别那些高度敏感的生物标志物基因,这些基因能够指示特定类型的癌症。在本研究中,我们提出了一种名为EODE的新方法,旨在解决癌症类型的分类问题,特别是在高维度且样本规模较小的基因表达谱情况下。EODE利用灰狼优化器(GWO)来优化特征子集,并构建一个经过优化的集成分类器。通过结合自然启发的特征选择和集成学习,EODE显著提高了模型的识别能力。
我们在涵盖多种癌症类型的35个数据集上进行了实验,结果表明,与四种自然启发的集成方法(PSOEL、EAEL、FESM和GA-Bagging-SVM)、六种基准机器学习算法(KNN、DT、ANN、SVM、DISCR和NB)、六种集成算法(RF、ADABOOST、RUSBOOST、SUBSPACE、TOTALBOOST和LPBOOST)以及七种自然启发的方法(ACO、CS、DE、GA、GWO、PSO和ABC)相比,我们的算法在分类准确性方面表现更好。
在未来的工作中,我们的目标是通过改进对多余和无效特征的筛选来提高算法的效率。此外,由于生物医学数据通常呈现出类别不平衡的特点,我们计划确保在类别不平衡的数据上获得稳健的结果。除了计算上的改进之外,我们还打算在来自不同临床队列的扩展基因表达数据集上评估所提出的方法。由于使用基因表达数据进行癌症亚型分型在指导个体化治疗决策方面具有巨大潜力,我们希望将这个计算流程转化到现实世界的临床设置中。
九、引用我们的工作
1
2
3
4
5
6
7
@article{wang2024eode,
title={Exhaustive Exploitation of Nature-inspired Computation for Cancer Screening in an Ensemble Manner},
author={Wang, Xubin and Wang, Yunhe and Ma, Zhiqiang and Wong, Ka-Chun and Li, Xiangtao},
journal={IEEE/ACM Transactions on Computational Biology and Bioinformatics},
year={2024},
publisher={IEEE}
}